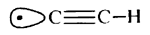
1: Materials and Molecular Simulations Center, Beckman Institute,
California Institute of Technology, Pasadena, CA 91125, USA
2: Xerox Palo Alto Research Center, 3333 Coyote Hill Road,
Palo Alto, CA 94304, USA
Nanotechnology 2 (1991)187-195.
This is a preliminary HTML version of the paper and might have errors.
Further information (links to related information and articles) is available at http://www.zyvex.com/nanotech/Habs/Habs.html.
Mechanical positioning of reactive species can be used to convert mechanical energy to chemical energy to select between alternative reactions, or to provide activation energy. Mechanosynthesis is the employment of these mechanochemical processes to synthesize molecular structures [1]. Atomically precise mechanosynthesis promises to let us manufacture complex systems of molecular machinery. Examples include: self-replicating assemblers [2], molecular-scale surgical systems [2], computers made with molecular logic elements [3] and macroscopic machines made of diamond-like materials. Construction of such systems will require the ability to manipulate structure precisely on an atomic level. The great specificity of the chemical reactions required to synthesize designs with specific atomic structures should be achievable with mechanochemical tools capable of positioning the reactive moieties with sub-angstrom accuracy. Mechanochemistry allows alternative reaction transition states to be selected by manoeuvring the reactive species into a position where the chosen reaction has the smallest barriers. Such positional control requires that the tool exert forces and torques on the reactive molecule to move it over the potential energy surface of interaction with the workpiece.
Applying positional control to reactions will require that the tool have certain properties to make synthesis reliable, feasible and practicable. The tool must (i) have the proper chemical properties, (ii) be relatively small to reduce steric interactions with the workpiece, (iii) be capable of remaining chemically and mechanically stable under thermal motions and strains induced during positioning, (iv) be bound to a system that can transfer forces and torques to the reactive portion of the tool, (v) be selective between alternative reactions and (vi) be easily made. Molecular tips attached to atomic force microscope (AFM) tips, scanning tunnelling microscope (STM) tips or molecular robotic arms have been suggested [4].
Because construction of atomically precise machinery might require about as many unit operations as there are atoms in the system, it is important that reactions be fast. To increase the speed of reactions with moderate barriers, forces can be exerted between the workpiece and reactive species to effectively increase the pressure on the system, reducing the barrier height. Moderate reductions in the barrier heights lead to substantial increases in the reaction rate because thermal vibrations have an exponential Boltzmann probability of overcoming the reaction barrier. Mechanochemistry not only reduces the barriers by converting mechanical energy to chemical energy, but also maximizes the effective reactive concentration by positioning the reactive moieties to best advantage. These speed-enhancing steps together with having many mechanochemical machines working simultaneously can compensate for the loss of parallelism of solution-based reactions. Mechanochemical synthesis also increases the range of synthetic steps that can be used to build novel structures by the use of applied torques; a moiety attached to both the tool and the workpiece can be twisted, for example, to break pi-bonds [1].
Nanomachines made of complex specific arrangements of diamond-like material offer several advantages. First, diamond is light and stiff. Macroscopic machines could be made stronger and simultaneously much lighter, making such activities as air and space travel substantially more practicable. Moving parts of such machines would be lighter and therefore faster. Furthermore, hydrocarbons are abundant, making raw materials readily available and inexpensive. Stiffness is not only a desirable property of finished machines, but it is also useful during construction since the material surrounding the reactive site on the workpiece must be stiff. This allows it to withstand the compressive forces that might be needed to reduce reaction barriers, to withstand the tensile forces during moiety abstraction and to withstand torques applied to break pi-bonds.
Building machines of diamond will include manoeuvring hydrocarbons into reactive sites, torsion of structures, insertions into bonds and preparation of reactive sites by removing unwanted moieties to create radical sites. Abstraction of hydrogen is likely to be the most repeated step and common to building a wide range of molecular structures, including diamond-like structures. Highly reactive species are commonly thought to play a crucial role in the chemical vapour deposition (CVD) synthesis of diamond [5-7]. The abstraction of hydrogen via any of several radicals is one of the central mechanisms involved in the growth of diamond. It is not unreasonable, therefore, to expect that the atomically precise synthesis of diamond-like materials will utilize site-specific hydrogen abstraction via a radical as one of the main steps. Drexler has proposed using a molecular tip made of an ethynyl radical [1]
|
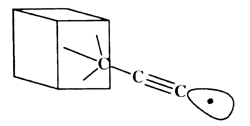
The C-H bond in acetylene is one of the strongest bonds to hydrogen; thus, the ethynyl radical formed by removing this hydrogen is likely to have a higher affinity for hydrogen than almost any other chemical structure. Further, the ethynyl radical can easily be incorporated into structures that provide a high degree of steric exposure. A structure resembling the propynyl radical, but with the carbon furthest from the radical site embed ded in an extended diamond-like structure (figure 1), provides both excellent steric exposure to the radical and a 'handle' for positioning the radical for the desired abstraction. Attachment of the tool to an STM or AFM tip may develop from technology designed for attachment of proteins to surfaces [4]. Drexler showed that the bending stiffness of an ethynyl-like tip attached to an adamantyl group is 6 N m-1 and can be increased to ~65 N m-1 by building up a surrounding collar [1]. If the reaction requires application of mechanical force to supplement thermal energy, then bending stiffness may need to be increased. Stiffness also is desirable to achieve selectivity. STM and AFM positioning is stable to more than sub-angstrom accuracy. However, bending modes of the ethynyl tip will be active at moderate temperatures. If during positioning of the tip, the bending of the radical and displacement of the AFM or STM relative to the work piece positions the reactive portion of the tip near a branched transition state, for example (one pathway leading to abstracting the neighbouring hydrogen), then selectivity is reduced. Drexler has shown that at worst at room temperature with a bending stiffness of 20 N m-1 and transition states separated by 1.2 Å the unwanted reaction rate is less than 10-12 times the rate of the target reaction [1]. Transition states between neighbouring hydrogens on the (111) surface of diamond are separated by 2.5 Å, and transition states for other possible reactions in diamond-like structures also generally exceed 1.2 Å, making mechanochemical reactions highly selective.
| R-H | D0 |
|---|---|
| H-H | 102.3 [25] |
| CH3-H | 105.1 [27] |
| (CH3)3C-H | 93.2 [27] |
| C6H5-H | 110.9 [27] |
| HCC-H | 126.6 [11], 131.3 [9] |
Several methods of creating the radical should be feasible. The process of creating the radical should take place in an inert environment: vacuum, helium, or some other extremely non-reactive system would be appropriate. The activation energy required to create the abstraction tool could be provided from thermal, mechanical, optical, or chemical sources. There are two obvious approaches. In the first, a precursor compound is activated to create the abstraction tool. The tool is then used once and discarded. A second precursor would then be activated to abstract a second hydrogen. Thus, in a functioning system using this approach, a steady supply of the precursor would be required as well as a method for disposing of the 'used' abstraction tools.
In the second approach, the abstraction tool would be refreshed by the removal of the hydrogen after each use. Of course, the ethynyl radical was selected on the basis of its strong C-H bond, so removal of the hydrogen might at first seem paradoxical. However, there are several methods of solving this problem. One would be first to weaken the C-H bond, and then abstract the hydrogen from the abstraction tool using a weaker radical. Drexler [1] proposed that the C-H bond could be weakened by positioning a weak radical near the carbon atom. A second weak radical could then abstract the hydrogen from the tip.
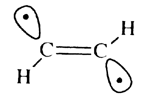
An alternative to the 'attack by two weak radicals' strategy would be photoexciting the acetylene to obtain the pi pi* state, which would rearrange to the structure
with a weak C-H bond and thereby allow removal of the hydrogen. Our primary concern is to analyse the energy barriers associated with hydrogen abstraction using alkynyl radicals to determine the feasibility of such a tool, rather than to analyse the methods of creating such a tool.
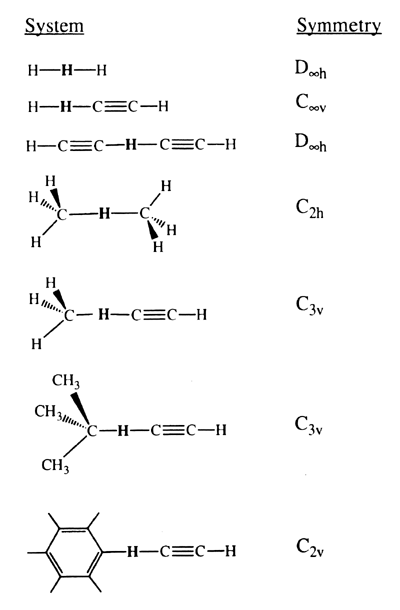
We model the chemically active site of the tool by the acetylene radical and determine the transition-state geometry and activation energy for transferring the hydrogen from several species: H2, CH4, C2H2, C6H6 and CH(CH3)3. The geometry of the various transition states can be used to position a working hydrogen abstraction tool for fast reaction and so as not to bend the tip. The barrier height itself can be used to calculate an abstraction rate at a given temperature and, thus, how long the abstraction tool must remain at the transition state until the probability that abstraction has occurred reaches a given value. Various levels of generalized valence-bond (GVB) and configuration-interaction (CI) ab initio calculations are used. To calibrate the accuracy of these calculations, we consider the abstraction barriers and transition states for hydrogen transfer between methyl and methane and for hydrogen transfer between H and H2 as compared to other theoretical and experimental results [13-15].
|
| R1-H-R2 | HF | GVBCI-SCF | CCCI | DCCI | HF*SD CI | GVB*SD CI |
| H-H-Ha | 24.3 | 9.9 | 10.3 | 10.3 | 10.5(9.6)c | 10.3(10.3) |
| CH3-H-CH3 | 34.9 | 27.8 | 29.8 | 17.5 | 22.5(19.5) | 20.4(18.8) |
| H-H-CCH | 11.6 | 8.0 | 5.4 | 0.8 | 4.5(3.2) | 3.3(2.7) |
| CH3-H-CCH | 14.6 | 8.6 | 10.2 | -2.9bb | 4.2(2.0) | 2.2(1.3) |
| (CH3)3C-H-CCH | 11.0 | 5.9 | 8.0 | -7.0 | 0.45(-2.78) | |
| C6H6-H-CCH | 18.3 | 12.0 | 14.3 | -0.7 | 7.7(4.1) | |
| HCC-H-CCH | 30.0 | 22.9 | 24.1 | 11.6 | 17.0(13.7) | 14.6(12.9) |
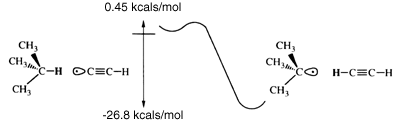
The barrier to the acetylene radical abstraction of hydrogen from isobutane (sp3 carbon) is conservatively estimated to be less than 0.45 kcal mol-1 (figure 2). Reaction barriers calculated at various levels of correlation are shown in table 2. The barrier to abstraction of hydrogen by the acetylene radical from benzene (sp2 carbon) is estimated to be less than 7.7 kcal mol-1. The Hartree-Fock times singles and doubles configuration interaction (HF*SD CI) consistently overestimates the generalized valence bond times singles and doubles configuration interaction (GVB*SD CI) barriers, while the dissociation-consistent configuration interaction (DCCI) consistently underestimates the GVB*SD CI barriers. The GVB*SD CI barriers will be conservatively high due to a lack of a third diffuse p function, zero-point corrections and lack of more correlation of the valence electrons. Transition states were optimized at the correlation-consistent configuration interaction (CCCI) level (table 3). Although CCCI does not accurately predict activation barriers, it does accurately describe the transition-state geometries. For the largest cases, the number of spin eigenfunctions in the configuration-interaction (CI) calculation grows beyond our computational capabilities (table 4). This makes abstraction from benzene and isobutane at the GVB*SD CI level impracticable; however, the overestimated, yet small, barriers at the HF*SD CI level show that the acetylene radical hydrogen abstraction is feasible, thermal vibrations at room temperature providing sufficient energy to overcome the barriers. Table 5 shows the exothermicities for the various abstractions. There is little difference in the accuracy of the methods in predicting the exothermicities because all the methods describe bound states rather well. Note that the exothermicities are for reactions where the product radical species are not allowed to relax. This describes abstraction from surfaces where relaxation is constrained. Exothermicities for gas-phase reactions will be higher. The transition state is poorly described by many semi-empirical methods and by ab initio methods with insufficient electron correlation and small basis sets, and large variation of the predicted barriers can be seen in table 2.
| R1-H-R2 | R1H bond length (Å) | R2H bond length (Å) | H-C-H anglea (deg) |
| H-H-H | 0.94 | 0.94 | |
| CH3-H-CH3 | 1.36 | 1.36 | 105.2 |
| H-H-CC-H | 0.80 | 1.61 | |
| CH3-H-CCH | 1.22 | 1.48 | 105.5 |
| (CH3)3C-H-CCH | 1.2 | 1.5 | |
| C6H5-H-CCH | 1.24 | 1.42 | |
| HCC-H-CCH | 1.28 | 1.28 |
| R1-H-R2 | CCCI | DCCI | HF*SD CI | GVB*SD CI |
| H-H-H | 191(386) | 191(386) | 139(278) | 191(386) |
| CH3-H-CH3 | 4914(9829) | 71666(143333) | 79428(158757) | 310778(621164) |
| H-H-CCH | 2160 | 18234 | 15189 | 53048 |
| CH3-H-CCH | 9477 | 150557 | 184851 | 737651 |
| (CH3)3C-H-CCH | 21676 | 695842 | 1675566 | |
| C6H5-H-CCH | 17265 | 587343 | 1514151 | |
| HCC-H-CCH | 2697(5398) | 42201(84406) | 55211(110314) | 221805(443182) |
| R1-H-R2 | HF | GVBCI-SCF | CCCI | DCCI | HF*SD CI | GVB*SD CI |
| H-H-CCH | 33.9 | 31.6 | 31.8 | 30.1 | 30.0(27.5)a | 29.9(28.1) |
| CH3-H-CCH | 28.9 | 28.4 | 27.0 | 29.2 | 26.9(25.5) | 26.8(25.4) |
| (CH3)3C-H-CCH | 28.5 | 27.9 | 24.2 | 27.4 | 26.8(25.9) | |
| C6H5-H-CCH | 18.7 | 18.2 | 15.4 | 18.9 | 18.3(18.0) |
Ideally, we would like to do a CI calculation in which all single and double excitations of the valence electrons are made into the virtual orbitals with reference to the three most important configurations describing the abstraction. This type of multi-reference CI (called a GVB*SD CI) has been well established in approximating results of complete CI calculations [17]. However, this CI has not been carried out for the largest cases, abstraction of hydrogen from isobutane and benzene by the acetylene radical. Thus we have considered some smaller CI calculations, which will do a good job in approximating the barriers for the larger CI. The first of these is the CCCI wavefunction [18, 19]. It involves making all single and double excitations of the active electrons and all single excitations of the other valence electrons into the virtual space relative to the three GVB references, or simply GVB*(SDactive + Svalence). The second CI, called a DCCI (dissociation-consistent configuration interaction), will add the double excitations, which are the product of a single excitation of an active electron and a single excitation of a valence electron, or GVB*(SDactive + Sactive*Svalence + Svalence). The third CI does all single and double excitations of the valence electrons (active and inactive) relative to only one reference, a calculation called HF*SD CI (or equivalently one-reference SDCI). Table 4 shows the sizes of the CI expansions in terms of the number of spin eigenfunctions (SEF) for each of the systems studied. The HF*SD CI already approaches the limits of our programs (about two million SEF) in the cases of isobutane and benzene, for which the GVB*SD CI is not possible. In all other cases, however, the GVB*SD CI are of small to medium size and will serve as benchmarks to calibrate the accuracy of the smaller CI.
The standard basis sets of Dunning and Huzinaga are used [20,21]. Their double-zeta contraction of the 9s5p set is used on all carbons, with the addition of one set of d polarization functions (Zd = 0.75). On the active carbons, diffuse s and p functions (Zs = 0.0474 and Zp = 0.0365) are also added. For active hydrogens or hydrogens bound to active carbons (in the case of methane), the triple-zeta contraction of the 6s set is used, supplemented with a p polarization function (Zp = 0.60). For all other hydrogens, the double-zeta contraction of the 4s set is used, scaled by a factor of 1.2.
 |
The basic geometries of the various systems studied are illustrated schematically in figure 3. The geometries will be optimized at the CCCI level. The orbital optimization at the GVBCI-SCF level is the most time-consuming step, so this CI will be a simple correction to that wavefunction. It would be impracticable to do the geometry optimization at a higher-level CI. It would also be impracticable to do a full geometry optimization, so certain constraints are assumed. Namely, only the parameters relevant to the description of the hydrogen abstraction (R1H and R2H or combinations thereof) will be optimized. In the case of abstraction from methane (by the methyl radical or by the acetylene radical), the H-C-Habs bond angle is also optimized since this angle changes from 109.5° for methane to 90° for the methyl radical. For isobutane, we would expect a small relaxation from a tetrahedral C-C-Habs bond angle to something more planar at the transition state and in the radical species. But Page and Brenner [22], in their work on abstraction of hydrogen from isobutane by atomic hydrogen, found that full relaxation of the t-butyl species reduced the abstraction barrier by only 1.7 kcal mol-1 at the GVBCVI-SCF level. GVBCI-SCF level. However, since the goal of this work is to show the feasibility of using an alkynyl radical tip as a hydrogen abstraction tool, a conservative overestimate of the abstraction barriers is acceptable. So the C-C-Habs bond angle is fixed to 109.5° in these calculations. All other radicals are expected to show little or no relaxation and are fixed to the experimental values of their hydrogen bound counterparts.
All calculations are run with the GVB [23] and MOLECULE-SWEDEN (an electronic structure program system written by Almlof et al [24]) suites of programs on the Caltech group's Alliant FX/80 and FPS5OO.
| HF | GVBCI-SCF | MR*SD CI | |
| Te(3Bu <-- 1E+g) | 72.3 | 92.0 | 90.2(87.6) |
| De(1E+g HCC-H) | 116.5 | 122.3 | 131.9(132.0) |
| De(3Bu HCC-H) | 44.2 | 30.3 | 41.7(44.4) |
Now that it has been established that an alkynyl-tipped hydrogen abstraction tool would be able to abstract hydrogen from diamond surfaces with little or no thermodynamic hindrances, it would be desirable to find a method for removing the hydrogen from the tip. A simple alternative, but less elegant strategy is to make a new tip for each abstraction and dispose of the tool after use. What makes the acetylene good at abstracting hydrogen is the strength of its C-H bond. However, this bond is quite weak in the 3Bu excited state (see table 6). We calculate a bond strength of 41.7 kcal mol-1 doing a GVBCI-SCF in which all 10 valence electrons are active in 10 orbitals, followed by a multi-reference times singles and doubles configuration interaction (MR*SD CI) in which all configurations in the GVBCI with coefficients < 0.05 are chosen as references. In the case of the triplet excited state, there are four references and, in the case of dissociated H + CCH, there are six references. The geometry for the excited state is optimized at the MR*SD CI level. The molecule is not linear in this state, having a C-C-H bond angle of 132.0°. The C-C bond length also increases to 1.38 Å from 1.20 Å for the ground state, reflecting the double bond character of this bond. The weakening of the C-C bond in the excited state leads directly to the weakening of the C-H bonds, as the triple bond character can be restored upon dissociation of one of the C-H bonds. The weakening of the C-H bond leaves the acetylene prone to abstraction, making it easy to remove the hydrogen and refresh the tip. So photoexcitation of the alkynyl tip from its ground state to the 1Bu excited state, followed by relaxation to the triplet would facilitate the breaking of the tip-hydrogen bond. Drexler also made an alternative proposal for removing the hydrogen from the tip [1].