Computational nanotechnology
by
Ralph C. Merkle
Xerox PARC
3333 Coyote Hill Road
Palo Alto, CA 94304
merkle@xerox.com
Copyright 1991 by Xerox Corporation.
All Rights Reserved.
This article was published in Nanotechnology, Volume 2,
1991, pages 134 through 141.
This version has been edited and updated for the web and
differs in some respects from the published version.
Nanotechnology is published by
IOPP (the Institute of
Physics Publishing).
Abstract
The major research objectives in
molecular
nanotechnogy are the
design, modeling, and fabrication of molecular machines and
molecular devices. While the ultimate objective must clearly be
economical fabrication, present capabilities preclude the
manufacture of any but the most rudimentary molecular structures.
The design and modeling of molecular machines is, however, quite
feasible with present technology. More to the point, such modeling
is a cheap and easy way to explore the truly wide range of molecular
machines that are possible, allowing the rapid evaluation and
elimination of obvious dead ends and the retention and more
intensive analysis of more promising designs. While it can be
debated exactly how long it will take to develop a broadly based
molecular manufacturing capability, it is clear that the right
computational support will substantially reduce the development
time. With appropriate molecular CAD software, molecular modeling
software (including available computational chemistry packages,
e.g., molecular mechanics, semi-empirical and ab initio programs)
and related tools, we can plan the development of molecular
manufacturing systems on a computer just as Boeing might "build"
and "fly" a new plane on a computer before actually manufacturing
it.
Introduction
The concept of a Universal Computer, a device able to compute
anything computable, dates back to
Babbage
in the early part of the
19th century[1], and to Turing, Church, and von Neumann in the
20th century[18].
The concept of a "Universal Constructor" is perhaps more recent. The
concept was well understood by von Neumann in the 1940's, who
defined a "Universal Constructor" in a two-dimensional cellular
automata world. Such a model is a mathematical abstraction
something like an infinite checkerboard, with several different
types of "checkers" that might be on each square. The different
pieces spontanesouly change and move about depending on what pieces
occupy neighboring squares, in accordance with a pre-defined set
of rules. Von Neumann used the concept of a Universal Constructor
in conjunction with a Universal Computer as the core components in
a self-replicating system[7]. The possibility of fabricating
structures by putting "...the atoms down where the chemist says..."
was recognized by Feynman in 1959[3]. Drexler recognized the
value of the "assembler" in 1977. (Note:
Drexler
said "Though assemblers will be powerful ... they will not be
able to build everything that could exist.")
The assembler is analogous to
von Neumann's Universal Constructor, but operates in the normal
three dimensional world and can build a large, atomically precise
structures by manipulating atoms and small clusters of atoms. He
published the concept in 1981[8] and in subsequent work [6] has
proposed increasingly detailed designs for such devices. The basic
design of Drexler's assembler consists of (1) a molecular computer,
(2) one or more molecular positioning devices (which might resemble
very small robotic arms), and (3) a well defined set of chemical
reactions (perhaps one or two dozen) that take place at the tip of
the arm and are able to fabricate a wide range of
structures using site-specific
chemical reactions.
It is now common for forecasts of future technical abilities to
include the ability to fabricate molecular devices and molecular
machines with atomic precision[4, 11, 19].
While there continues to be debate about the exact time frame, it
is becoming increasingly accepted that we will, eventually, develop
the ability to economically fabricate a truly wide range of
structures with atomic precision. This will be of major economic
value. Most obviously a molecular manufacturing capability will
be a prerequisite to the construction of molecular logic devices.
The continuation of present trends in computer hardware depends on
the ability to fabricate ever smaller and ever more precise logic
devices at ever decreasing costs. The limit of this trend is the
ability to fabricate molecular logic devices and to connect them
in complex patterns at the molecular level. The manufacturing
technology needed will, almost of necessity, be able to
economically manufacture large structures (computers) with atomic
precision (molecular logic elements). This capability will also
permit the economical manufacture of materials with properties that
border on the limits imposed by natural law. The strength of
materials, in particular, will approach or even exceed that of
diamond. Given the broad range of manufactured products that
devote substantial mass to load-bearing members, such a development
by itself will have a significant impact. A broad range of other
manufactured products will also benefit from a manufacturing
process that offers atomic precision at low cost.
Given the promise of such remarkably high payoffs it is natural to
ask exactly what such systems will look like, exactly how they will
work, and exactly how we will go about building them. One might
also enquire as to the reasons for confidence that such an
enterprise is feasible, and why one should further expect that our
current understanding of chemistry and physics (embodied in a
number of computational chemistry packages) should be sufficient
to explain the operating principles of such systems.
It is here that the value of computational nanotechnology can be
most clearly seen. Molecular machine proposals, provided that they
are specified in atomic detail (and are of a size that can be dealt
with by current software and hardware), can be modeled using the
tools of computational chemistry.
There are two modeling techniques of particular utility. The first
is molecular mechanics, which utilizes empirical force fields to
model the forces acting between nuclei[9, 10, 15, 16, 17]. The
second is higher order ab initio calculations, which will be
discussed in a few paragraphs.
A few links to internet computational chemistry resources
are provided here to provide those new to the area with some
feel for what's available.
Molecular Mechanics
Molecular mechanics allows computational modeling of the positions
and trajectories of the nuclei of individual atoms without an undue
computational load. Current packages available on personal
computers can readily do energy minimizations
on systems with thousands of
atoms, while supercomputers can handle systems with hundreds of
thousands of atoms or more. More complex analyses, particularly
analyses that involve searching through large configuration
spaces, can limit the size of system that can be effectively handled.
As will be discussed later the need to search through large
configuration spaces (as when determining the native folded
structure of an arbitrary protein) can be avoided by
the use of relatively
rigid structures (which differ from relatively floppy proteins
and have few possible configurations).
The modeling of machine components in vacuum reduces the need
to model solvation effects, which can also involve significant
computational effort.
In molecular mechanics the individual nuclei are usually treated
as point masses. While quantum mechanics dictates that
there must be a certain degree of positional
uncertainty associated with each nucleus, this positional
uncertainty is normally significantly smaller than the typical
internuclear distance. Bowen and Allinger[16] provide a good
recent overview of the subject.
While the nuclei can reasonably be approximated as point masses,
the electron cloud must be dealt with in quantum mechanical terms.
However, if we are content to know only the positions of the nuclei
and are willing to forego a detailed understanding of the
electronic structure, then we can effectively eliminate the quantum
mechanics. For example, the H2 molecule involves two nuclei.
While it would be possible to solve Schrodinger's equation to
determine the wave function for the electrons, if we are content
simply to know the potential energy contributed by the electrons
(and do not enquire about the electron distribution) then we need
only know the electronic energy as a function of the distance
between the nuclei. That is, in many systems the only significant
impact that the electrons have on nuclear position is to make a
contribution to the potential energy E of the system. In the case
of H2, E is a simple function of the
internuclear distance r. The function E(r) summarizes and replaces
the more complex and more difficult to determine wave function for
the electrons, as well as taking into account the inter-nuclear
repulsion and the interactions between the electrons and the
nuclei. The two hydrogen nuclei will adopt a position that
minimizes E(r). As r becomes larger, the potential energy of the
system increases and the nuclei experience a restoring force that
returns them to their original distance. Similarly, as r becomes
smaller and the two nuclei are pushed closer together, we also find
that a restoring force pushes them farther apart, again restoring
them to an equilibrium distance.
More generally, if we know the positions
r1, r2, .... rN of N
nuclei, then
E(r1, r2, .... rN)
gives the potential energy of the
system. Knowing the potential energy as a function of the nuclear
positions, we can readily determine the forces acting on the
individual nuclei and therefore can compute the evolution of their
position over time. The function E is a newtonian potential energy
function (not quantum mechanical), despite the fact that the
particular value of E at a particular point could be computed from
Schrodinger's equation. That is, the potential energy E is a
newtonian concept, but the particular values of E at particular
points are determined by Schrodinger's equation.
Often called the Born-Oppenheimer approximation[13,20], this approach
allows a great conceptual and practical simplification in the
modeling of molecular systems. While it would in principle be
possible to determine E by solving Schrodinger's equation, in
practice it is usual to use available experimental data and to
infer the nature and shape of E by interpolation. This approach,
in which empirically derived potential energy functions are created
by interpolation from experimental data, has spawned a wide range
of potential energy functions,
many of which are sold commercially.
Because the gradient of the potential energy function E defines a
conservative force field F, molecular mechanics methods are also
called "force field" methods. While it is common to refer to
"empirical force field" methods, the more recent use of ab initio
methods to provide data points to aid in the design of the force
fields[17] makes this term somewhat inaccurate, though still widely
used.
The utility of molecular mechanics depends crucially on the
development of accurate force fields. Good quality force fields
have been developed for a fairly broad range of compounds including
many compounds of interest in biochemistry[9, 10, 15, 16, 17].
While we will not attempt to survey the wide range of force fields
that are available, one particular subset of compounds for which
good quality force fields are available involve H, C, N, O, F, Si,
P, S, Cl (and perhaps a few others) when they are restricted to
form chemically uncomplicated structures (e.g., bond strain is not
too great, dangling bonds are few or absent, etc). Many atomically
precise structures which should be useful in nanotechnology fall
in this class and can be modeled with an accuracy adequate to
determine the behavior of molecular machines.
 A
potential energy function of particular utility in modeling
diamondoid structures was described by
Brenner (Empirical potential for hydrocarbons for use
in simulating the chemical vapor deposition of diamond films,
Donald
W. Brenner, Phys Rev B, Vol. 42, No. 15, November 15 1990,
pages 9458-9471). Brenner's potential energy function, though
limited to the elements carbon and hydrogen, has the great advantage
that it will handle transition states, unstable structures,
and the like. Thus, given essentially arbitrary coordinates of
the carbon and hydrogen atoms in a system, Brenner's potential
will return the energy of the system. This permits molecular
dynamical modeling of arbitrary hydrocarbon systems,
including systems which use synthetic reactions
involved in the synthesis of diamond.
A particular reaction of interest is
the selective abstraction of a chosen hydrogen atom
from a diamond surface. See Surface patterning
by atomically-controlled chemical forces: molecular dynamics
simulations by Sinnott et al., Surface Science 316 (1994),
L1055-L1060; see also the work of
Robertson et al. for an illustration of the use of
Brenner's potential in modeling the behavior of graphitic gears,
including failure modes).
Brenner has commented on the utility of this potential for modeling
proposed molecular machine systems.
A
potential energy function of particular utility in modeling
diamondoid structures was described by
Brenner (Empirical potential for hydrocarbons for use
in simulating the chemical vapor deposition of diamond films,
Donald
W. Brenner, Phys Rev B, Vol. 42, No. 15, November 15 1990,
pages 9458-9471). Brenner's potential energy function, though
limited to the elements carbon and hydrogen, has the great advantage
that it will handle transition states, unstable structures,
and the like. Thus, given essentially arbitrary coordinates of
the carbon and hydrogen atoms in a system, Brenner's potential
will return the energy of the system. This permits molecular
dynamical modeling of arbitrary hydrocarbon systems,
including systems which use synthetic reactions
involved in the synthesis of diamond.
A particular reaction of interest is
the selective abstraction of a chosen hydrogen atom
from a diamond surface. See Surface patterning
by atomically-controlled chemical forces: molecular dynamics
simulations by Sinnott et al., Surface Science 316 (1994),
L1055-L1060; see also the work of
Robertson et al. for an illustration of the use of
Brenner's potential in modeling the behavior of graphitic gears,
including failure modes).
Brenner has commented on the utility of this potential for modeling
proposed molecular machine systems.
Of course, the "accuracy" of the force fields depends on the
application. A force field which was accurate to (say) 10 kcal/
mole would be unable to correctly predict many properties of
interest in biochemistry. For example, such a force field would
lead to serious errors in predicting the correct three-dimensional
structure of a protein. Given the linear sequence of amino acids
in a protein, the protein folding problem is to determine how it
will fold in three dimensions when put in solution. Often, the
correct configuration will have an energy that differs from other
(incorrect) configurations by a relatively modest amount, and so a
force field of high accuracy is required.
Consider, however, the bearing illustrated in figures 1 and 2.
Unlike the protein folding problem, where an astronomical range of
configurations of similar energy are feasible, the bearing
basically has only one configuration: a bearing. While the protein
has many unconstrained torsions, there are no unconstrained
torsions in the bearing. To significantly change any torsion angle
in the bearing would involve ripping apart bonds. Thus, the same
force field which is of marginal utility in dealing with strands
of floppy protein is quite adequate for solid blocks of stiff
diamondoid material. Not only will small errors in the force
field still result in an accurate prediction of the global minima,
(which in this case will be a single large basin in the potential
energy surface) but also the range of possible structures is so
sharply limited that little or no computational effort need be
spent comparing the energies of different configurations. Long
computational runs to evaluate the statistical properties of
ensembles of configurations are thus eliminated.

Figure 1. A molecular bearing.
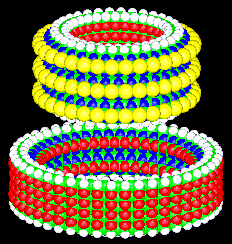
Figure 2. The bearing taken apart.
This style of design has been called, only half in jest, "molecular
bridge building" because bridges are also designed with large
safety margins.
This observation, that the same force field that is inadequate for
one class of structures is quite adequate for the design and
modeling of a different class of structures, leads to a more
general principle. Computational experiments generally provide an
answer with some error distribution. If the errors produced by the
model are of a similar size to the errors that would result in
incorrect device function, then the model is unreliable. On the
other hand, if the errors in the model are small compared with the
errors that will produce incorrect device function, then the
results of the model are likely to be reliable. If a device design
falls in the former category, i.e., small errors in the model will
produce conflicting forecasts about device function, then the
conservative course of action is to reject the proposed design and
keep looking. That is, we can deliberately design structures
which are indeed adequately handled by our computational tools.
As illustrated by the bearing, there are a wide range of
basically mechanical structures whose stability is sufficiently
clear and whose interactions with other structures is sufficiently
simple that their behavior can be adequately modeled with currently
available force fields.
An even stronger (although somewhat more subtle) statement is
possible. The bearing illustrated in figures 1 and 2 is simply a
single bearing from a very large class of bearings. The strain in
the axle and the sleeve is proportional to the diameter of the
bearing. By increasing the diameter, we can reduce the strain.
Thus, we can design bearings in this broad class in which the strain
can be reduced to whatever level we desire. Because we are dealing
with what amounts to a strained block of diamond, the fact that we
can reduce strain arbitrarily means that we can design a bearing
whose local structure bears as close a resemblance to an unstrained
block of diamond as might be wished. We can therefore be very
confident indeed that some member of this class will perform the
desired function (that of a bearing) and will work in accordance
with our expectations. Given that we have developed software tools
that are capable of creating and modeling most of the members of a
broad class of devices, then we can investigate many individual
class members. This investigation then lets us make rather
confident statements about the functionality that members of this
class can provide, even if there might be residual doubts about
individual class members.
A moments reflection will show that the class of objects which are
chemically reasonably inert, relatively stiff (no free torsions),
and which interact via simple repulsive forces that occur on
contact; can describe a truly vast class of machines. Indeed, it
is possible to design computers, robotic arms and a wide range of
other devices using molecular parts drawn from this class[1,2,5,6].
Ab Initio Methods
While empirical force fields are sufficiently accurate to model the
behavior of chemically stable stiff structures interacting with
other chemically stable stiff structures, they do not (at present)
provide sufficient accuracy to deal with chemical transitions.
Thus, if we wish to model the manufacture of a molecular part (such
as the axle or sleeve of the bearing of figures 1 and 2) then we
must use higher order ab initio techniques (another paper in this
issue illustrates what is meant by this in detail[14]). These
techniques impose severe constraints on the number of atoms that
can be modeled (perhaps one or two dozen heavy atoms, depending on the
hardware, software, and specific type of modeling being attempted),
but can provide an accuracy sufficient to analyze the chemical
reactions that must necessarily take place during the synthesis of
large, atomically precise structures.
In [14], an analysis of the
abstraction of a hydrogen atom from various structures, including
isobutane (which serves as a model of the diamond (111) surface),
has been carried out to illustrate the kind of reactions that are
of interest. More generally, higher order ab initio techniques are
sufficient to analyze the addition or removal of a small number of
atoms from a specific site on a work piece. Synthesis of a large
object would then consist of repeated site specific applications
of a small number of basic operations, where each basic operation
changed the chemical structure of only a small number of atoms at
a time. Provided that we reject reaction mechanisms where the
result predicted depends on errors that are smaller than can
reasonably be modeled, the analysis of these basic operations can
be satisfactorily carried out with current methods and hardware.
Several reactions mechanisms have been discussed, and a great many
more are feasible.
Sufficiency of Current Modeling Methods
In summary, it is quite possible to adequately model the behavior
of molecular machines that satisfy two constraints: (1) they are
built from parts that are sufficiently stable that small errors in
the empirical force fields do not raise significant questions about
the shape or stability of the parts, and (2) the synthesis of the
parts is done by using positionally controlled reactions, where the
actual chemical reactions involve a relatively small number of
atoms whose behavior can be adequately modeled with higher order
ab initio methods.
Clearly, not all molecular machines satisfy these constraints. Is
the range of molecular machines which do satisfy these constraints
sufficiently large to justify the effort of designing and modeling
them? And in particular, can we satisfactorily model Drexler's
assembler within these constraints?
The fundamental purpose of an assembler is to position atoms. To
this end, it is imperative that we have models that let us determine
atomic positions, and this is precisely what molecular mechanics
provides. Robotic arms or other positioning devices are basically
mechanical in nature, and will allow us to position molecular parts
during the assembly process. Molecular mechanics provides us with
an excellent tool for modeling the behavior of such devices.
The second fundamental requirement is the ability to make and break
bonds at specific sites. While molecular mechanics provides an
excellent tool for telling us where the tip of the assembler arm
is located, current force fields are not adequate to model the
specific chemical reactions that must then take place at the
tip/work-piece interface involved in building an atomically precise
part (though this statement must be modified in light of the work
done with Brenner's potential, discussed above).
For this, higher order ab initio calculations are
sufficient.
The glaring omission in this discussion is the modeling of the kind
of electronic behavior that occurs in switching devices. Clearly,
it is possible to model electronic behavior with some degree of
accuracy, and equally clearly molecular machines that are basically
electronic in nature will be extremely useful. It will therefore
be desirable to extend the range of computational models discussed
here to include such devices. For the moment, however, the
relatively modest inclusion of electrostatic motors as a power
source is probably sufficient to provide us with adequate "design
space" to design and model an assembler. While it might at first
glance appear that electronics will be required for the
computational element in the assembler, in fact molecular
mechanical logic elements will be sufficient. Babbage's proposal
from the 1800's clearly demonstrates the feasibility of mechanical
computation[1] (it is interesting to note that a working model of
Babbage's
difference engine has been built and is
on display at the British Museum of Science.
They were careful to use parts machined
no more accurately than the parts available to Babbage, in order
to demonstrate that his ideas could have been implemented in the
1800's). Drexler's analysis of a specific molecular mechanical
logic element[2] and further analysis of system design issues[6]
also make it clear that molecular mechanical computation is
sufficient for the molecular computer required in an assembler.
Molecular electronic proposals for computation have not been worked
out as clearly at the system level as the molecular mechanical
concepts[6]. Thus, at the moment, molecular mechanical proposals
are better understood, at least in this particular context. This
situation is likely to change, and when it does the electronic
designs could be incorporated (where appropriate) into the design
and modeling of an assembler. However, it is not entirely obvious
that electronic designs will prove superior to molecular mechanical
designs, particularly when device parameters such as size and
energy dissipation are considered. While it seems virtually
certain that electronic devices will prove faster than molecular
mechanical devices, it is less obvious that electronic devices must
necessarily be either smaller or dissipate less energy[5] (though
confer more recent work on
reversible logic).
Indeed,
given the difficulty of localizing individual electrons, it seems
possible that electronic devices will prove to be inherently larger
than molecular mechanical devices. This might provide a long term
role for molecular mechanical devices as high density memory
elements, or as logic elements when space (or atom count) is
particularly constrained.
Whether the assembler is designed and built with an electronic or
mechanical computer is less significant than designing and building
it. By way of example, a stored program computer can be built in
many different ways. Vacuum tubes, transistors, moving mechanical
parts, fluidics, and other methods are all entirely feasible.
Delaying the development of the Eniac because transistors are
better than vacuum tubes would have been a most unwise course of
action. Similarly, as we consider the design, modeling, and
construction of an assembler, we should not hesitate to use simpler
methods that we can understand in favor of better methods that are
not yet fully in hand.
The methods of computational chemistry available today allow us to
model a wide range of molecular machines with an accuracy
sufficient in many cases to determine how well they will work. We
can deliberately confine ourselves to the subset of devices where
our modeling tools are good enough to give us confidence that the
proposals should work. This range includes bearings, computers,
robotic arms, site-specific chemical reactions utilized in the
synthesis of complex structures, and more complex systems built up
from these and related components. Drexler's assembler and related
devices can be modeled using our current approaches and methods.
Molecular Compilers
(Much of this section has been superceded by work on molecular
CAD tools with graphical user interfaces: in particular, work
by Geoff Leach on Crystal Clear.)
Computational nanotechnology includes not only the tools and
techniques required to model proposed molecular machines, it must
also include the tools required to specify such machines.
Molecular machine proposals that would require millions or even
billions of atoms have been made. The total atom count of an
assember might be roughly a billion atoms. While commerically
available molecular modeling packages provide facilities to
specify arbitrary structures, it is usually necessary to "point and
click" for each atom involved. This is obviously unattractive for
a device as complex as an assembler with its roughly one billion
atoms. It should be clear that molecular CAD tools will be required
if we are to specify such complex structures in atomic detail with
a reasonable amount of effort. An essential development is that
of "molecular compilers" which accept, as input, a high level
description of an object and produce, as output, the atomic
coordinates, atom types, and bonding structure of the object.
A simple molecular compiler has already been written at PARC, and
was used to produce the bearing of figures 1 and 2. The
specification of the axle was:
scale 0.9
tube 0 0.75 0.75 1.75 0 0 0 17 -17 0 5.25 5.25
grid 0 0.5 -0.5 0 1 1
delete 1.25 0 0
grid 0 0.25 0 0 0 0.25
change O_3 to S_3 1.5 0 0
The first line, "scale 0.9," is simply an instruction to shrink the
size of the axle by 10% compared with the normal size. This allows
the axle to be positioned inside the sleeve before joint
minimization of the axle and sleeve is done (using MSI's PolyGraf,
a commercially available molecular mechanics package that
implements both MM2[9] and the Dreiding II force field[10]). If
the axle w were not done, then the atoms in the axle and the sleeve
would be comingled, and minimization would produce meaningless
results.
The second line begins with the "tube" specifier. This tells the
program to produce a tubular structure, rather than a "block" or a
"ring." the "ring" specifier is used to produce toroidal
structures, e.g., tubes that have been further bent into donuts.
The first triplet of numbers following "tube," "0 0.75 0.75,"
specifies the offset to be used for the crystal lattice. The second
triplet, "1.75 0 0" specifies that the surface of the tube is (100),
and that the thickness of the tube wall is 1.75 lattice spacings.
The third triplet, "0 17 -17," specifies the direction and length
of the circumference of the tube. The direction [0 17 -17] (or [0
1 -1]) is at right angles to [1 0 0], the direction of the tube
surface. The circumference of the tube is simply the length of the
[0 17 -17] vector. Finally, "0 5.25 5.25" gives the direction of
the axis and the length of the tube.
This does not fully specify the axle of the bearing, for the 100
surface has been cut circumferentially to produce grooves. The
next command, "grid 0 0.5 -0.5 0 1 1" specifies that a grid is to
be laid down on the surface. The grid is specified by two vectors,
"0 0.5 -0.5" and "0 1 1,"" which give the directions and lengths
of the edges of the unit parallelogram from which the grid is
composed. The next command, "delete 1.25 0 0"" specifies that the
point at coordinates "1.25 0 0" is to be deleted, and further, that
all points in the same location with respect to any unit
parallelogram of the grid are also to be deleted. Thus, this "grid
.... delete ...." cuts out the grooves that are visible on the
outer surface of the axle.
Finally, the commands "grid 0 0.25 0 0 0 0.25," and "change O_3
to S_3 1.5 0 0" are used to lay down a new, very fine grid that
causes all oxygen atoms on the surface of the axle to be changed
to sulfur.
A similar set of commands was used to specify the sleeve of the
bearing.
It is easy to specify bearings with different surfaces, surface
orientations, circumferences, lengths, etc. To select a (111),
(110), (312), or any other surface, it is sufficient to change the
vector that specifies the surface, and the vectors that specify the
tangent to the surface and the axis of the tube (which must be at
right angles to the surface vector). Thus, complex structures
involving many atoms can be generated quickly and easily with a few
lines of input specification.
The C source code is available at URL
ftp://ftp.parc.xerox.com/pub/nano/tube.c.
The program will generate output in either PolyGraf
format or in Brookhaven format, so the output should be readable
by most computational chemistry packages.
The software required to design and model complex molecular
machines is either already available, or can be readily developed
over the next few years. Many of the modeling issues can be dealt
with by existing commercially available computational chemistry
packages. The molecular compilers and other molecular CAD tools
needed for this work can be implemented using generally understood
techniques and methods from computer science. Using this approach,
it will be possible to substantially reduce the development time
for complex molecular machines, including Drexler's assembler.
This approach is similar in spirit to the computer aided design and
modeling used to speed the development of many products today.
The author was part of a general purpose computer start up which
successfully designed and built a new computer from scratch. This
included the hardware, software, compilers, operating system, etc.
During this process, extensive use was made of computational models
to verify each level of the design. The operating system was
written in Pascal, and checked out on another computer. The
compilers were written in Pascal, and also checked out on another
computer. The code produced by the compilers was checked out on
an instruction set simulator. The microcode was checked out on a
micro-instruction simulator. The logic design was checked out with
logic level simulation tools, and circuit simulation packages were
used to verify the detailed electronic design. All this work was
done at the same time. The software was written and debugged even
though the machine on which it would eventually be executed didn't
exist. The microcode was written and debugged before the hardware
was available. When the hardware was finally made available,
system integration went relatively rapidly.
Imagine, for a moment, how long it would have taken to develop this
system had we carried out the development in the obvious sequential
fashion. First, we would have implemented the hardware and only
then begun work on the microengine. Later, with the hardware and
microengine fully checked out and working, we could have developed
and debugged the microcode. With this firmly in hand, we could
then have written the compilers and verified the code they
produced. Finally, we could have implemented and checked out the
operating system. Needless to say, such a strategy would have been
very slow and tedious.
Doing things in the simple and most obvious way often takes a lot
longer than is needed. If we were to approach the design and
construction of an assembler using the simple serial method, it
would take a great deal longer than if we systematically attacked
and simultanesouly solved the problems that arise at all levels of
the design at one and the same time. That is, by using methods
similar to those used to design a modern computer, including
intensive computational modeling of individual components and sub-
systems, we can greatly shorten the time required to design and
build complex molecular machines.
This can be further illustrated by considering the traditional
manner of growth of our synthetic capability over time (figure 3).
Today, we find we are able to synthesize a certain range of
compounds and structures. As time goes by, we will be able to
synthesize an ever larger range of structures. This growth in our
ability will proceed on a broad front, and reflects the efforts of
a broad range of researchers who are each pursuing individual goals
without concern about the larger picture into which they might fit.
As illustrated in figure 4, given sufficient time we will
eventually be able to synthesize complex molecular machines simply
because we will eventually develop the ability to synthesize just
about anything.
However, if we wish to develop a particular kind of device, e.g.,
an assembler, then we can speed the process up (as illustrated in
figure 5) by conducting computational experiments designed to
clarify the objective and to specify more precisely the path from
our current range of synthetic capabilities to the objective. Such
computational experiments are inexpensive, can provide very
detailed information, are possible for any structure (whether we
can or cannot synthesize it) and will, in general, reduce the "time
to market" for the selected product.
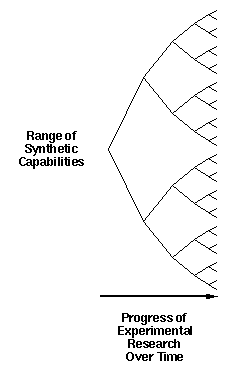
Figure 3. Experimental advances in our synthetic capabilities
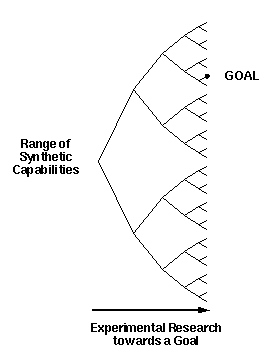
Figure 4. Experimental progress towards a goal
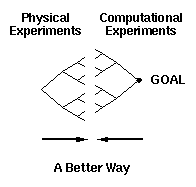
Figure 5. A more efficient method of achieving a goal
Computational experiments let us examine structures quickly and
easily, rejecting those which have obvious defects (a precursor to
the bearing shown in figure 1, for example, was too strained. By
modifying the design and again minimizing the structure, we found
a design with an acceptable strain). This kind of examination of
the "design space" is impossible with physical experiments today,
but is easily done with computational experiments.
Computational experiments also provide more information. For
example, molecular dynamics can literally provide information
about the position of each individual atom over time, information
which would usually be inaccessible in a physical experiment.
Of course, the major advantage of computational experiments over
physical experiments in the current context is the simple fact that
physical experiments aren't possible for molecular machines that
we can't make with today's technology. By using computational
models derived from the wealth of experimental data that is
available today, we can (within certain accuracy bounds) describe
the behavior of proposed systems that we plan to build in the
future. If we deliberately design systems that are sufficiently
robust that we are confident they will work regardless of the small
errors that must be incurred in the modeling process, we can design
systems today that we will not be able to build for some years, and
yet still have reasonable confidence that they will work.
By fully utilizing the experience that has been developed in the
rapid design and development of complex systems we can dramatically
reduce the development time for molecular manufacturing systems.
It is possible to debate how long it will be before we achieve a
robust molecular manufacturing capability. However, it is very
clear that we'll get there sooner if we develop and make
intelligent use of molecular design tools and computational models.
These will let us design and check the blueprints for the new
molecular manufacturing technologies that we now see on the
horizon, and will let us chart a more rapid and more certain path
to their development.
Acknowledgements
The author would like to thank Rick Danheiser and Eric Drexler for
their very helpful conversations, as well as the anonymous referees
of this paper who contributed many good suggestions and ideas. The
author, of course, remains responsible for any defects in the
finished product.
References
- 1.) Of the Analytical Engine, by
Charles Babbage, 1864, in:
Perspectives on the Computer Revolution, by Zenon W. Pylyshyn,
Prentice Hall, 1970.
- 2.) Rod Logic and Thermal Noise in the Mechanical
Nanocomputer, by K. Eric Drexler, in Proceedings of the 3rd
International Symposium on Molecular Electronic Devices, F.L.
Carter, R.E. Siatkowski, H. Wohltjen, eds., North-Holland 1988.
- 3.) There's Plenty of Room at the Bottom, a
talk by Richard Feynman at an annual meeting of the American
Physical Society given on December 29, 1959.
- 4.) A Small Revolution Gets Under Way, by Robert Pool,
Science, Jan. 5 1990.
- 5.) Two Types of
Mechanical Reversible Logic, by Ralph C. Merkle.
- 6.) Nanotechnology: molecular machinery,
manufacturing, and computation, by K. Eric Drexler, Wiley 1992.
- 7.) Theory of Self-Reproducing Automata, by John von
Neumann, edited and completed by Arthur W. Burks, University of
Illinois Press, 1966.
- 8.) Molecular engineering: An approach to the development of
general capabilities for molecular manipulation, by K. Eric
Drexler, Proceedings of the National Academy of Sciences U.S.A.
78(9): 5275-5278, 1981.
- 9.) Molecular Mechanics, by Ulrich Burkert and Norman L.
Allinger, ACS Monograph 177, American Chemical Society, 1982.
- 10.) DREIDING: A Generic Force Field For Molecular
Simulations, by Stephen L. Mayo, Barry D. Olafson, and William
A. Goddard III, The Journal of Physical Chemistry, 1990, 94, pages
8897-8909.
- 11.) New Frontiers in Computing and Telecommunications, by
J. A. Armstrong, IBM Vice President for Science and Technology and
Chief Scientist, Creativity, June 1991, Vol 10 No. 2, pages 1-6.
- 13.) Methods of Molecular Quantum Mechanics, by R. McWeeny,
Second Edition, Academic Press 1989.
- 14.) Theoretical studies of a hydrogen abstraction tool for
nanotechnology, by Charles B. Musgrave, Jason K. Perry, Ralph
C. Merkle, and William A. Goddard III, Nanotechnology.
- 15.) Reviews in Computational Chemistry, Vol 2, Kenny B.
Lipkowitz and Donald B. Boyd, VCH Publishers, 1991.
- 16.) Molecular Mechanics: The Art and Science of
Parameterization, by J. Phillip Bowen and Norman L. Allinger,
page 81-97 of reference [15].
- 17.) New Approaches to Empirical Force Fields, by Uri Dinur
and Arnold T. Hagler, pages 99-164, reference [15].
- 18.) Perspectives on the Computer Revolution, Zenon W.
Pylyshyn, Prentice Hall 1970.
- 19.) Atom by Atom, Scientists Build `Invisible' Machines of the
Future, by Andrew Pollack, The New York Times Science Section,
Tuesday, November 26, 1991, page B7.
- 20.) The Nuclear Motion Problem in Molecular Physics, by
B. T. Sutcliffe, in Advances in Quantum Chemistry, Vol. 28,
1997, pages 65-80. (This reference added 97.08.31).
 This
page is part of the
nanotechnology web site.
This
page is part of the
nanotechnology web site.