Theoretical studies of diamond mechanosynthesis
reactions.
Stephen P. Walch
Thermosciences
Institute
NASA Ames Research
Center
Moffett Field, CA
94035-1000
and
Ralph C. Merkle
Xerox, PARC
3333 Coyote Hill Road
Palo Alto, CA 94304
This paper is available on the
web at http://www.zyvex.com/nanotech/mechanosynthesis.html.
It was published in Nanotechnology
9 (1998) pages 285-296. The published version differs in some respects
from this web page.
Abstract
Density functional
theory methods have been used to examine the interaction of i) the carbene and
C2 tools with a pair of radical sites on the diamond (111) surface
and ii) the carbene tool with a surface dimer on the reconstructed diamond
(100) surface.
For the (111) surface,
the carbene tool (carbenecyclopropene) is found to bond preferentially to a
single radical site (on top site) rather than at a bridged site. This means
this tool is not useful for adding a carbon to diamond (111). The C2
tool, on the other hand, is found to add a bridged C2 molecule,
through a series of steps which are overall exothermic. The carbene tool can
add a carbon to the bridged C2 molecule, leading to a bridged C3
molecule perpendicular to the surface, by an overall exothermic series of
steps. If another radical site is activated, the C3 can bend over to
a three fold coordinated position, with only a small barrier. Thus, this series
of steps can be used to create a three fold coordinated C3 molecule
on the diamond (111) surface.
For the surface dimer
on the reconstructed (100) surface, the carbene tool is found to add with no
barrier if the angle between the tool and the surface is allowed to vary or
with a 0.09 aJ (13 kcal/mol) barrier for a C2v constrained approach.
In this case, a bridged site is strongly favored, and the subsequent steps of
sequentially breaking the p and s bonds between the tool and added carbon atom
are all feasable. Thus, this series of steps can add a bridged C atom to the
reconstructed diamond (100) surface.
I. Introduction.
Manufactured products are made by arranging atoms. Current manufacturing methods provide
limited control over the position of individual atoms in the final product. A manufacturing technology able to precisely
arrange atoms in patterns chosen to enhance material properties and
capabilities, rather than in the limited range of patterns that current
manufacturing methods allow, would permit revolutionary improvements in product
performance and capabilities. A
material which in theory provides particularly attractive capabilities is
diamond. Its advantages for electronic
applications have been known for some time (Geis and Angus, 1992). Its
strength-to-weight ratio is over 50 times that of metals used in aerospace
applications and would permit major performance improvements in such
applications (McKendree, 1996). One
defect of diamond as a structural material (in addition to high manufacturing
cost with current technology, which has been addressed by other authors
(Drexler, 1992; Merkle, 1994) and will not be considered here) is it's inherent
brittleness. However, modified structures related to diamond should retain most
of the high strength while not being brittle. Since it isn't yet possible to
make molecularly precise materials with todays technology, theory and modelling
can play an important role in evaluating the properties of proposed new
materials.
Conventional methods (e.g., CVD (Celii
and Butler, 1991)) are unable to synthesize diamond with the precision
envisioned here. As no immediately
experimentally accessible approach appears able to achieve this objective, the
use of theoretical methods and in particular quantum chemistry methods become
indispensable if we are to examine and evaluate proposals. A general proposal advanced by Drexler
(Drexler, 1992) is to use mechanical forces to position atoms and molecules in
precise locations using a small robotic arm. Drexler proposed some molecular
tools along with a few specific reactions utilizing those tools which could be
used to synthesize diamond and related structures. One such tool was the
carbene insertion tool (Fig. 1a), which Drexler suggested could be used on the
diamond (110) surface. Application of the carbene tool to the (100) surface
also appears plausible. The hypothesis that this tool might be useful on the
(111) surface has not been investigated before, and interaction of the carbene
tool with the (111) and reconstructed (100) surfaces are examined here. In
addition, we examine the interaction of the C2 tool with the (111)
surface.
A hypothetical application of the carbene
insertion tool to the (111) surface is
shown in a sequence of steps for adding a C atom to the diamond (111) surface,
illustrated in Fig. 2. The first step (Fig. 2a-b) is use of a hydrogen
abstraction tool ( an alkynyl radical) (Musgrave, Perry, Merkle, and Goddard,
1991) to abstract two adjacent surface
H atoms leading to two adjacent radical sites ( referred to as dangling bonds).
The second step (Fig. 2b-c) is an insertion of the carbene tool leading to a
bridged structure. The last step (Fig. 2c-d) is application of torque to break
the CC p bond followed by application of force to break the remaining
single bond between the carbene carbon and the cyclopropene ring, leaving a
bridged carbon atom behind on the surface.
Theory can answer a number of
questions related to the reactions in Fig. 2. A key issue is the nature of the
interaction between a carbene and two adjacent dangling bonds on the surface.
The dangling bonds on the surface are separated by 0.252 nm (2.52 Å) compared
to a CC distance of 0.121 nm (1.21 Å) in acetylene, and it is not clear if for
this CC distance it is energetically more favorable for the carbene to bridge
accross two surface carbon atoms or to bond to just one surface carbon atom. If
the latter is more favorable, then there are no restraining forces to permit
breaking the CC p bond by application of torque, and subsequent
application of force would simply reverse the addition process. If, on the
other hand, bridging is favored, it is well established that carbene insertion
proceeds by a pathway involving edge on approach, and it is not clear what the
barrier would be for addition in the Woodward-Hoffman forbidden perpendicular
approach as shown in Fig. 2.
Fig. 3 shows two reactions for vinylidene
(CCH2). The first ( Fig. 3a) is addition of the carbene pair to a p
orbital of acetylene. This is analagous to to the pathway above leading to a
bridged carbene on the surface. The second reaction (Fig. 3b) is the addition
of a radical (in this case H atom) to vinylidene leading to vinyl radical. This
is analagous to the pathway above leading to bonding to a single surface carbon
atom. The vinylidene plus acetylene reaction will be used as a calibration for
the DFT method since there are accurate ab initio calculations available for
this system (Walch and Taylor, 1995).
In this paper we use density functional
theory methods to investigate the interaction of several mechanosynthesis tools
with the diamond (111) and reconstructed (100) surfaces using a cluster model.
II. Computational Details.
The calculations were carried out with the
density functional theory(DFT) method with the Becke-3LYP functional and the
6-31G basis set using the program system G94. The clusters for the (111)
surface are frozen at the bulk diamond geometry For the reconstructed (100)
surface, the first layer is allowed to relax [ (2x1) dimer reconstruction) ],
but the deeper layers are frozen at the bulk geometry. The first and second
nearest neighbor distances are taken as 0.1545 nm (1.545 Å) and 0.2522 nm
(2.522 Å), respectively.
III. Discussion.
a. Cluster model for diamond (111).
Fig. 4 shows the two clusters which were
used. The larger cluster has three carbon atoms in the surface layer and six
carbon atoms in the second layer. These 9 carbon atoms are located at the
positions appropriate to the unreconstructed diamond (111) surface. The bonds
between the second layer carbons and the other carbons, which are not included
in the cluster, are simulated by CH bonds, where the CH bond has a bond length
of 0.11 nm (1.1 Å) and is directed toward the appropriate bulk carbon atom. The
smaller cluster has two surface carbon atoms and one second layer carbon atom
with the bonds to the remaining bulk carbons simulated by CH bonds as was done
for the larger cluster. The larger cluster has three surface dangling bonds and
the smaller cluster has two surface dangling bonds. In the calculations the
dangling bonds which are not involved in the reaction are also bonded to an H
atom. In the following the larger cluster is denoted by cluster and the smaller
cluster is denoted by cluster2.
An important question is the amount of
interaction between two surface dangling bonds. This was quantified by
computing the overlap between the two radical orbitals in a GVB(pp) calculation
(for cluster2). The overlap is found to be only 0.016 and the triplet state is
found to be 0.03 aJ (3.7 kcal/mol) below the singlet state. This result
indicates that the dangling bonds may be thought of as localized radicals with
only weak interaction with adjacent dangling bonds.
b. Cluster model for reconstructed diamond
(100).
The unreconstructed diamond (100) surface
has rows of surface carbon atoms which are carbene like, i.e they have two
bonds to the second layer and two unsatisfied valences. Relaxation of the
surface layer leads to a reconstructed surface with rows of dimers. Fig. 5
shows a cluster for one surface dimer on the diamond (100) surface. This
cluster has two surface, four second layer, two third layer, and one fourth
layer carbon atoms, respectively. The atoms in the second through fourth layers
were held at the bulk positions and the dangling bonds are tied off with H
atoms. The two surface layer atoms were allowed to relax in a density
functional theory (DFT) calculation. In the dimer there is a radical orbital on
each C atom, and these orbitals can be combined into singlet or triplet spin
states. The simplest picture is that both states have a CC s bond
and the singlet state has a weak p bond as well. An important question, which has
been considered in the literature (Weiner, Skokov, and Frenklach, 1995.) is the
amount of p bonding in the dimer. This may be quantified in
various ways; perhaps the best way is to compute the overlap between the two
radical orbitals in a GVB(pp) calculation. First the dimer geometries were computed
for the singlet and triplet spin states using DFT. This gives a CC bond length
of 0.1421 nm (1.421 Å) for the singlet and 0.1714 nm (1.714 Å) for the triplet.
At the DFT singlet geometry, a GVB calculation gives an overlap of the radical
orbitals of 0.462. These properties indicate a weak p bond.
Another indication of the amount of p bonding is the singlet --> triplet
excitation energy which is computed to be about 0.07 aJ (10 kcal/mol).
c. Structure of the carbene tool.
The carbene tool, shown in Fig. 1, was
simulated by the case where the two R groups are H (C4H2).
One question that arose was the relative energetics and barrier to
isomerization between the three membered ring (carbenecyclopropene) and four
membered ring ( 1,2-dehydro-cyclobutadiene) forms of C4H2.
This question could be answered by high level ab-initio theory. Calculations
using the CASSCF/ ICCI method for structures optimized at the CASSCF level and
including zero-point effects estimated from the harmonic frequencies show that
the carbene cyclopropene isomer is more stable by 0.20 aJ (29.4 kcal/mol). (See
Table I. and Fig. 6) Thus, isomerization to the four membered ring form is
unlikely at low temperatures. One reason for the low stability of
1,2-dehydro-cyclobutadiene is that the in plane p bond, that can be
formally drawn, is very weak because of the approximately 90° CCC angle.
(Compare to the surface dimer on the reconstructed diamond (100) surface
discussed in section IIIb.)
The carbenecyclopropene form of the carbene
tool has a singlet ground state. The lowest triplet state has the two radical
orbitals in plane (a1 and b2 in C2v symmetry)
and is computed to be 0.31 aJ (44.3 kcal/mol) higher at the CASSCF level (0.30
aJ (42.6 kcal/mol) for DFT). The location of this triplet state is important,
since the C2v constrained insertion into a p bond
(e.g. of a surface dimer on the reconstructed (100) surface) involves crossing
of the surface arising from the ground states of the tool and cluster with the
surface arising form the triplet states of the tool and cluster.
d. Structure of the C2 tool.
Fig. 7 shows an isomerization pathway
for the C2 tool. The energetics are obtained with the CASSCF/ ICCI
method as was done for the carbene tool. (See Table II.) Here it is seen that
the carbene form ( 5 membered ring) is more stable than the six membered ring
form by 0.10 aJ (15.1 kcal/mol). However, there is a barrier of 0.11 aJ (16.2
kcal/mol) between the two forms. Thus, the six membered ring form, which is the
C2 tool, is a stable minimum on the potential energy surface, and
should have a useful lifetime at low temperatures. As for the carbene tool, we
are also interested in the location of the triplet state of this tool. CASSCF
finds the singlet state to be 0.09 aJ (10.3 kcal/mol) below the triplet state.
This is about the same as for a surface dimer on the reconstructed (100)
surface, and is consistent with an activated CC p bond.
e. Comparison of energetics for vinylidene plus
acetylene reaction.
Calibration calculations have been
carried out for several systems using both DFT and ab-initio methods. Table III
shows energetics for several structures in the addition of vinylidene to
acetylene ( Fig. 3a). The entrance channel barrier (sp1) is found to be 0.01 aJ
(1.7 kcal/mol) compared to 0.04 aJ (5.4 kcal/mol) for ab initio. The structure
min2 is found to be 0.40 aJ (58.0 kcal/mol) below reactants compared to 0.37 aJ
(53.6 kcal/mol) for ab initio. Since the ab-initio values use double-zeta
quality basis sets it is expected that the ab initio values would become closer
to the DFT values if larger basis sets were used; i.e. the barrier height
should decrease and the binding energy of min2 with respect to reactants should
increase. Thus, the comparison here suggests that the DFT method reproduces the
ab inito results well enough to be used for the calculations planned here,
which seek to answer semi-quantitative issues.
f. Energetics for CH3 + vinylidene.
The vinylidene plus acetylene reaction
serves as a model for two adjacent surface radical sites (dangling bonds) which
are close enough to have a significant amount of singlet pairing (this is
appropriate e.g. for dimer sites on the reconstructed diamond (100) surface).
In the opposite extreme, where the adjacent radical sites are too far apart to
interact significantly (diamond (111) surface), the interaction with a carbene
is like the interaction of a simple radical with a carbene. One example of such
an interaction is the reaction of methyl radical (CH3) with
vinylidene (CCH2) (Fig. 3b). Table IV shows energetics for this
reaction at the ICCI level of theory. Here it is seen that the barrier for
addition of the CH3 radical to the carbene pair of vinylidene is
only 0.002 aJ (0.3 kcal/mol). Thus, this process is predicted to be more facile
than insertion into a CC p bond in acetylene.
g. Addition of the carbene tool (C4H2)
to the diamond (111) surface.
Table V shows energetics for bonding the
carbene tool to the two cluster models of diamond (111). Min1 is obtained by
bonding the carbene tool to a cluster with only one radical site. Thus, this
forces the system to go to a structure like Fig. 3b, arising by addition of a
radical orbital to the carbene pair of the carbene tool (on top site). Min2 is
the bridged site over two adjacent radical sites. Finally, min3 is a structure
like min1 except that there is also an adjacent radical site. From Table V and
Fig. 8. It is seen that the on top site is favored over the bridged site for
diamond (111). It is also seen that the binding energies for min1 and min3 are
very similar, which indicates that the adjacent radical site is only weakly
interacting with the carbene tool bonded at an on top site. In Fig. 8, a
barrier is shown separating min2 and min3. The saddle point for this process
was not found. Min2 was obtained as a singlet state, while min3 was obtained as
a triplet state. This is reasonable, since for min3 the triplet state should be
nearly degenerate with the open shell singlet state. The failure to find a
saddle point connecting min2 and min3 on the singlet surface is not surprising,
since for an ab initio wavefunction this would involve a complex recoupling of
the electron spins, which is a process which may not be well described by DFT.
These results indicate that the process depicted in Fig. 2 is not
likely to work for diamond (111), since for the favored on top site there would
be no restraining forces to oppose the applied torque to break the CC p bond.
From comparison of the energetics with the two clusters, it is seen that the
smaller cluster (cluster2) gives results in semiquantitative agreement with
those obtained with the larger cluster. This is consistent with the rapid
convergence with cluster size typically seen for covalently bound clusters.
h. Addition of the C2 tool (C4H2O2)
to diamond (111).
The C2tool (Fig. 1b) may be
thoght of as a C2 molecule bonded to glyoxal. As discussed in
section IIId, bonding to glyoxal considerably activates the in plane CC p bond, since
the OCCO framework is strongly bent leading to in plane radical orbitals, which
are directed at approx 120° angles with respect to the CC bond. Thus, the
overlap between these radical orbitals is low (comparable to the dimer on the
reconstructed (100) surface, where the overlap of the two p
orbitals is 0.462). The C2v constrained approach of the C2
tool to a pair of adjacent radical orbitals on the (111) surface is a
Woodward-Hoffman forbidden process, which involves the crossing of the ground
state surface with the surface arising from the triplet states of the tool and
the cluster. In this case, the triplet state of the tool is 0.07 aJ (10
kcal/mol) above the singlet, while the triplet state of the surface is 0.03 aJ
(4 kcal/mol) below the singlet state.
Thus, the excitation energy to the upper surface is 0.04 aJ (6 kcal/mol), at
infinite separation. Combining these considerations, addition of the C2
tool accross two surface dimers on the (111) surface is expected to be a
barrier-less process. This is what the calculations show, and the lack of a
barrier is depicted in Fig. 9 as a straight line going down to the complex. The
binding energy between the C2 tool and the surface is computed to be nearly
1.39 aJ (200 kcal/mol) ( 1.39 aJ (199.8 kcal/mol) and 1.30 aJ (187.4 kcal/mol)
with cluster2 and cluster, respectively). (See Table VI.) The dissociation
process in which a glyoxal comes off and leaves a C2 behind has only
a slight barrier with respect to products ( about 0.01 aJ (1 kcal/mol) ). As
expected, the glyoxal has to tip out of plane in order to dissociate to the
ground state. Overall the reaction is exothermic by about 0.69 aJ (100
kcal/mol) ( 0.69 aJ (98.9 kcal/mol) and 0.63 aJ (90.5 kcal/mol) with cluster2
and cluster, respectively). These calculations indicate that the C2
tool will be effective in adding a bridged C2 species to the diamond
(111) surface.
i. Diamond (111) + bridged C2 plus
carbene tool.
Fig. 10 and Table VII show the
interaction between a C2 bridged on diamond (111) and the carbene
tool. In this case bridged bonding is favored over bonding to one C of the
bridged C2. This reflects a situation in which the two surface
radical orbitals, which correspond to the two electrons of the bent CC p bond,
are more strongly interacting than in the case of adjacent radical sites on
diamond (111). As for the results shown
in Fig. 8, min2 was calculated as a singlet while min3 was calculated as a
triplet. Fig. 11 and Table VII also show the energetics for the process
cluster2 + C2 plus carbene tool going to cluster2 + C3 + cyclo-C3H2.
Here min1 is the bridged structure depicted in Figs. 7 and 8 and is bound by
0.68 aJ (97.5 kcal/mol). Min3 is the case where the carbene tool bonds to one C
of the bridged C2 as depicted in Fig. 7 and is bound by
0.62 aJ (88.8 kcal/mol) Min2 is the structure where the cyclopropene
moity is twisted 90° such that the CC p bond is broken. This is still bound
by 0.30 aJ (43.4 kcal/mol) with respect to reactants. Finally, the
products, a bridged C3 and cyclo-C3H2, are bound by 0.22aJ (31.1 kcal/mol) with
respect to reactants. Thus, these calculations suggest that use of the C2
tool followed by use of the carbene tool is a viable reaction sequence to put a
C3 on the diamond (111) surface.
j. Rearangement of chemisorbed C3.
The previous sequence resulted in a C3
unit sitting perpendicular to the diamond (111) surface. Fig. 12 and Table VIII
show the energetics for a reaction pathway leading to a cyclic C3
unit bridged across a three fold site on diamond (111). The barrier to this
process is only 0.06 aJ (8.4 kcal/mol).
k. Addition of the carbene tool to a surface
dimer on the reconstructed diamond (100) surface.
As discussed in section IIIb, a dimer on
the reconstructed (100) surface has an activated p bond, but still has a
significant amount of p bonding. Thus, the addition of a carbene (such
as the C2 tool) to this surface has some of the features of the
addition of vinylidene to acetylene, which is depicted in Fig. 3a. (See also
Walch and Taylor, 1994.) The lowest energy pathway is shown in Fig. 13. Here it
is seen that this is a somewhat typical carbene insertion pathway, in that the
carbene approaches in a parallel orientation to the CC p bond.
One feature which is peculiar to this system is that the carbene approaches to
the outside of the CC p bond. This approach results from the
approximately 120° angles which the GVB orbitals of the p bond
make with the CC bond. From Fig. 13 it is seen that the pathway initially is
dominated by shortening of the forming CC bond, but then switches to mainly an
angular change to give ultimately a bridged carbene structure. This pathway has
no barrier.
The C2v constrained approach
is expected to have a barrier. The upper state in the curve crossing leading to
this barrier arises from the triplet states of the carbene tool and the surface
dimer. In this case the triplet state of the surface dimer is 0.07 aJ (10
kcal/mol) above the singlet state, and the triplet state of the carbene tool is
0.30 aJ (43 kcal/mol) above the singlet state. Thus, the excited state surface
is about 0.37 aJ (53 kcal/mol) above the ground state surface at infinite
separation. The calculations show a barrier of about 0.09 aJ (13 kcal/mol)
arising from this surface crossing. These two pathways are shown in Fig. 14 and
the energies are given in Table IX. Here it is seen that the bridged carbene
tool is bonded to the surface by about 118 kcal/mol. Twisting the C3H2
moitey by 90° such that the CC p bond is broken costs 0.45 aJ (65.1 kcal/mol),
while the remaining single bond is worth 0.51 aJ (73.5 kcal/mol). This is less
than the energy of a bridged carbon on the surface dimer ( about 0.83 aJ (120
kcal/mol) for the carbene tool) so the CC bond between the added carbon and the
departing C3H2 is expected to break in preference to the
bond of the bridged carbon to the surface.
IV. Conclusions.
Density functional theory methods have been used
to examine the interaction of the carbene and C2 tools with a pair of
radical sites on the diamond (111) surface and of the carbene tool with a dimer
on the reconstructed diamond (100) surface. Calibration calculations for the
reaction of vinylidene with acetylene, which is analagous to some of the
systems studied here, indicate that the DFT method gives reasonable results.
On diamond (111), the carbene tool
(carbenecyclopropene) is found to bond preferentially to a single radical site
(on top site) rather than at a bridged site. This result is believed to arise
because of the large separation ( 0.252 nm (2.52 Å) ) and resulting weak
interaction between adjacent radical sites on the diamond (111) surface. For
the on top site there would be no opposing forces to permit breaking the CC p bond
between the added carbon and the cyclopropene moity, and the carbene tool would
not useful for adding a carbon to diamond (111).
The C2 tool, on the other
hand, is found to add a bridged C2 molecule to the diamond (111)
surface through a series of steps which are overall exothermic. The carbene
tool can add a carbon to the bridged C2 molecule, leading to a
bridged C3 molecule perpendicular to the surface, once again by
a series of steps which are exothermic.
If another radical site is activated, the C3 can bend over to a
three fold coordinated position, with only a small barrier. Thus, this series
of steps can be used to create a three fold coordinated C3 molecule
on the diamond (111) surface.
The carbene tool adds to a surface dimer
on the rearranged (100) surface preferentially by a typical carbene insertion
pathway involving a nearly parallel approach of the reactants (barrierless
process). A C2v constrained approach has a barrier of about 0.09 aJ
(13 kcal/mol) This suggests that a mechanosynthesis process in which the C2v
constrained approach is used would encounter some significant torques on the
carbene tool. The series of steps proposed (Drexler, 1992) for use of the
carbene tool is found to be reasonable.
These calculations demonstrate the utility
of the theory in determining which proposed mechanosynthesis reactions will be
useful.
References:
Celii, F. G. and
Butler, J. E. 1991 Annu Rev. Phys. Chem.
42 643-684.
Drexler, K. E 1992 Nanosystems: Molecular Machinery,
Manufacturing, and Computation, Wiley.
Geis, M.W., and Angus,
J.C. 1992 Scientific American October 84.
Merkle, R. C. 1994 Self replicating systems and low cost
manufacturing in The Ultimate Limits
of Fabrication and Measurement, M.E. Welland, J.K. Gimzewski, eds.; Kluwer,
Dordrecht 25-32.
Musgrave C. B., Perry,
J. K., Merkle, R. C., and Goddard, W. A. Nanotechnology
1991 2 187-195.
Walch, S. P. and
Taylor, P. R 1994 J. Chem. Phys. 103 4975-4979.
Weiner, B, Skokov, S,
and Frenchlach, M 1995 J. Chem. Phys. 102 5486-5491.
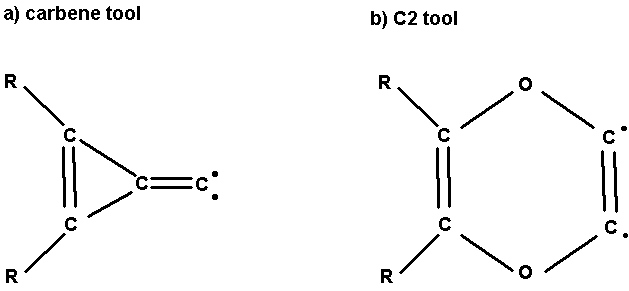
Fig. 1. The carbene tool and C2 tool.
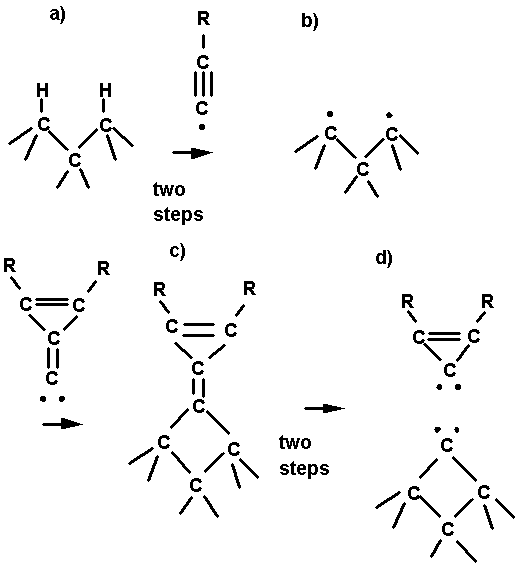
Fig. 2. A possible reaction sequence for adding
a bridged carbon atom to the diamond (111) surface. The first step is
abstraction of H atoms from two adjacent surface carbon atoms leading to
adjacent radical sites. The next step is insertion of the carbene end of the C2
tool leading to a bridged structure.
The last step is application of torsion to break the CC p bond
followed by application of force to break the remaining CC s bond.
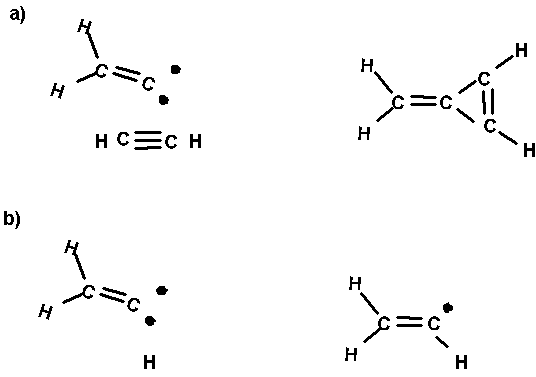
Fig. 3. Two reactions of vinylidene. a) reaction
with acetylene to give a cyclopropene structure. b) reaction with H atom to give vinyl radical. These
reactions are prototypes for addition of the carbene tool to give bridged and
on top sites, respectively.
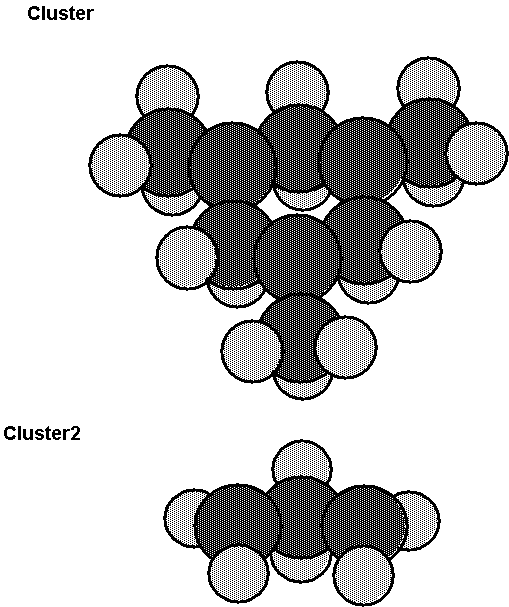
Fig. 4. Two clusters which were used to model
the diamond (111) surface. The larger cluster (denoted as cluster) has three
surface carbon atoms, while the smaller cluster (denoted as cluster2) has two
surface carbon atoms. The atoms are fixed at the locations for the
unreconstructed surface.
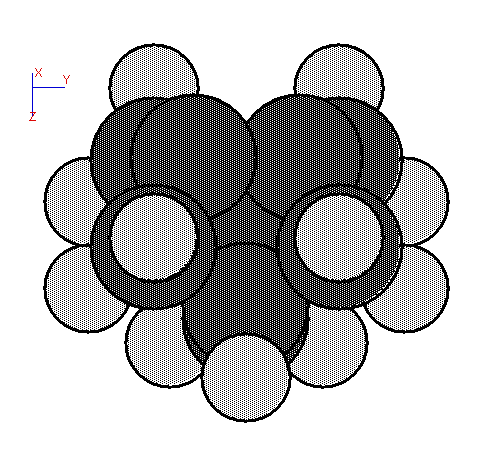
Fig. 5. Cluster for a dimer on diamond (100).
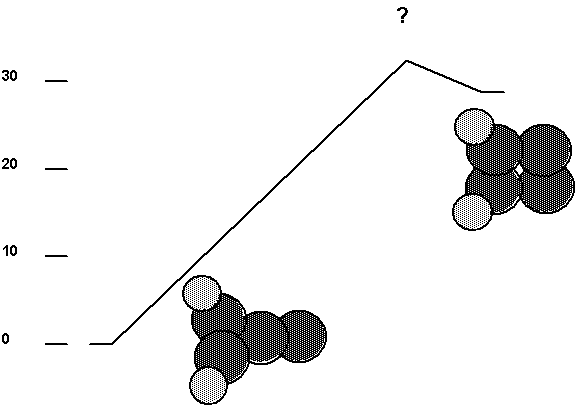
Fig. 6. Isomerization pathway for the carbene
tool. The barrier height has not been determined but is greater than the
endothermicity. The ordinate is energy in kcal/mol (one kcal/mol=0.00694 aJ).
The same energy units are used in the remaining figures.
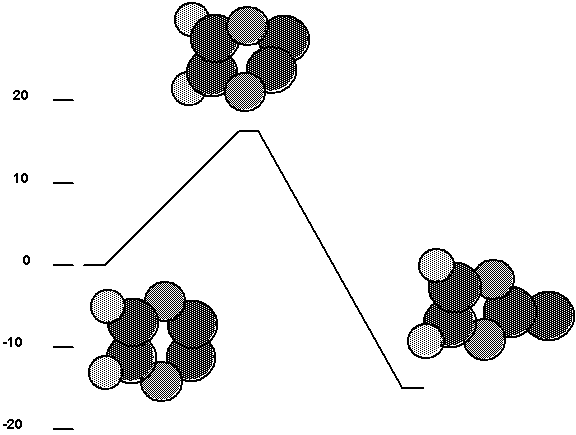
Fig. 7. Isomerization pathway for the C2
tool.
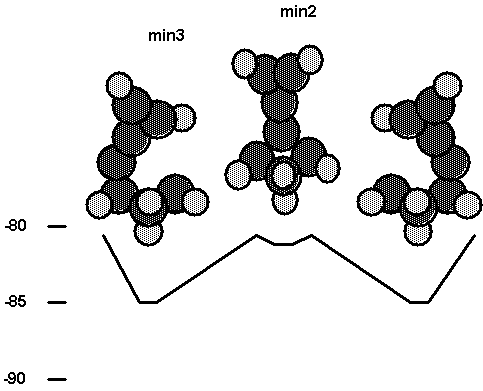
Fig. 8. Energetics for the interaction of the
carbene tool with cluster2.
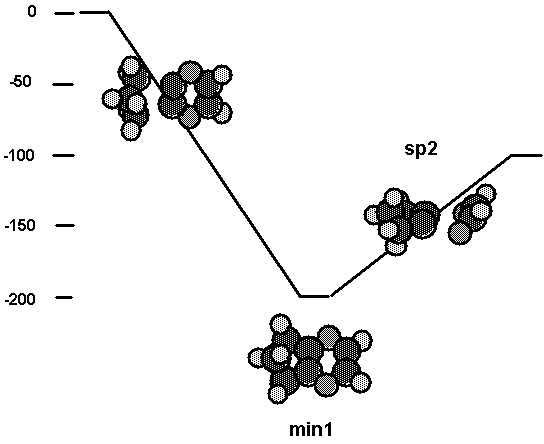
Fig. 9. Energetics for interaction of the C2
tool with cluster2.
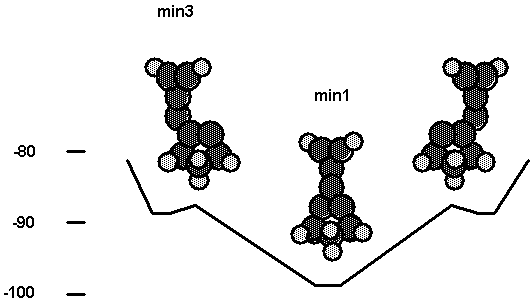
Fig. 10. Energetics for the interaction of
the C2 tool with a C2
molecule bridged across two radical sites on cluster2.
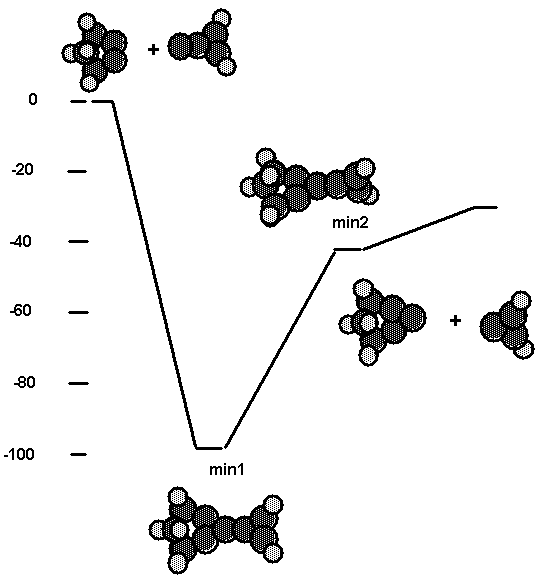
Fig. 11. Energetics for the reaction of the C2
tool with a C2 molecule bridged across two radical sites on
cluster2. The product is a C3 molecule sitting perpendicular to the
surface.
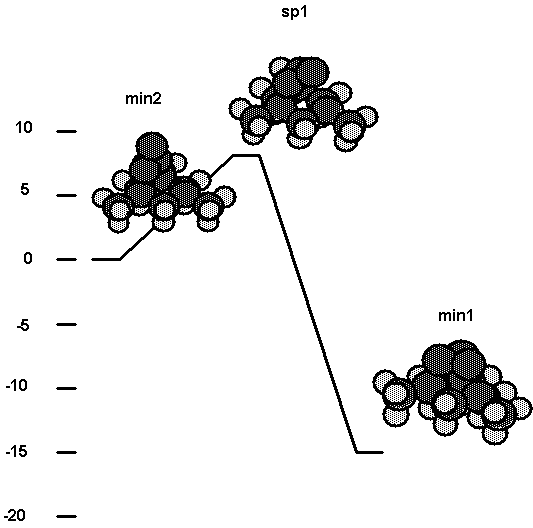
Fig. 12. Energetics for a bridged C3
molecule interacting with an adjacent radical site. The product is a three fold
coordinated C3 molecule.

Fig. 13. Minimum energy pathway for insertion of
the carbene tool into a surface dimer on the reconstructed diamond (100)
surface. The energies (in kcal/mol) with respect to carbene tool plus the
cluster are also given.
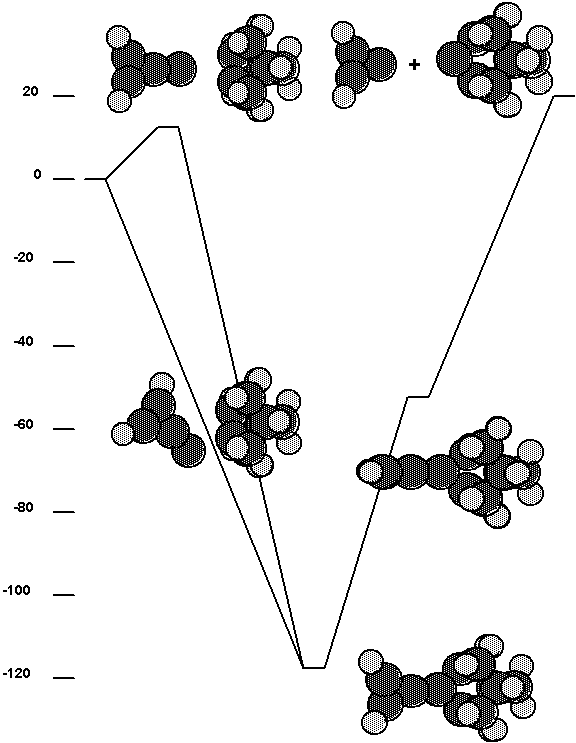
Fig. 14. Energetics for the c2 tool plus a
surface dimer on the reconstructed diamond (100) surface.
Table I. Energetics for
isomers of C4H2. (carbene tool)
|
structure |
ICCI(ICCI
+Q +152.)a |
ZPEb |
DEc |
|
carbenecyclopropene |
-152.90656(-.96332) |
.036514 |
0.0 |
|
1,2-dehydrocyclobutadiene |
-152.85853(-.91750) |
.037702 |
0.20 |
a ICCI is internally contracted CI, ICCI +Q is
internally contracted CI plus a multireference Davidson's correction.
b zero-point vibrational energy estimated from
the CASSCF harmonic frequencies.
c Energy difference (aJ) including correction for
zero-point energy.
d. Unless indicated otherwise all energies are
in EH ( one EH =4.359 748 2 x 10-18 J). The
same convention is used for the remaining tables.
Table II. Energetics
for isomers of C4O2H2. (C2 tool)
|
structure |
ICCI(ICCI+Q+
302.) |
ZPE |
DEa |
|
min1 |
-302.79852(-0.89419) |
.051756 |
0.0 |
|
sp |
-302.76198(-0.86500) |
.048373 |
0.11 |
|
min2 |
-302.82217(-0.91752) |
.051101 |
-0.10 |
a Energy difference (aJ) including correction for
zero-point
Table III. Energetics
for vinylidene + acetylene.
|
|
DFT |
|
|
Ab
Initio |
|
structure |
Energy |
ZPEa |
DEb |
DEb |
|
acetylene |
-77.31201 |
.02724 |
|
|
|
vinylidene |
-77.24135 |
.02437 |
|
|
|
|
-154.55336 |
.05161 |
0.0 |
0.0 |
|
|
|
|
|
|
|
sp1 |
-154.55368 |
.05462 |
0.01 |
0.04 |
|
|
|
|
|
|
|
min2 |
-154.65561 |
.06137 |
-0.40 |
-0.37 |
a zero-point vibrational energy estimated from
the DFT harmonic frequencies.
b Energy difference (aJ) including correction for
zero-point
Table IV. Energetics
for CH3 + vinylidene (pDZ optimized structures).
|
structure |
ICCI(ICCI
+Q +116.)a |
ZPEb |
DEc |
|
|
|
|
|
|
CH3
+ CCH2 |
-116.73157(-.77535) |
.055247 |
0.0 |
|
CH3-CCH2
SP |
-116.73373(-.77897) |
.059325 |
0.002 |
a ICCI is internally contracted CI, ICCI +Q is
internally contracted CI plus a multireference Davidson's correction.
b zero-point vibrational energy estimated from
the CASSCF harmonic frequencies.
c Energy difference (aJ) including correction for
zero-point
Table Va. Cluster2 + C4H2
Energetics.
|
structure |
DFT |
DEa |
|
cluster2
+ H |
-118.43037 |
|
|
C4H2 |
-153.32306 |
|
|
|
-271.75343 |
0.0 |
|
|
|
|
|
min1 |
-271.88691 |
-0.58 |
|
|
|
|
|
cluster2 |
-117.74505 |
|
|
C4H2 |
-153.32306 |
|
|
|
-271.06811 |
0.0 |
|
|
|
|
|
min2 |
-271.19816 |
-0.57 |
|
min3 |
-271.20344 |
-0.59 |
a Energy difference (aJ).
Table Vb. Cluster + C4H2
Energetics.
|
structure |
DFT |
DEa |
|
cluster
+2H |
-353.05775 |
|
|
C4H2 |
-153.32306 |
|
|
|
-506.50457 |
0.0 |
|
|
|
|
|
min1 |
-506.504568 |
-0.54 |
|
|
|
|
|
cluster
+ H |
-352.38179 |
|
|
C4H2 |
-153.32306 |
|
|
|
-505.70485 |
0.0 |
|
|
|
|
|
min2 |
-505.81816 |
-0.49 |
|
min3 |
-505.83054 |
-0.55 |
a Energy difference (aJ).
Table VIa. Cluster2
+ C2tool Energetics.
|
structure |
DFT |
DEa |
|
cluster2 |
-117.74505 |
|
|
c2tool |
-303.70708 |
|
|
|
-421.45213 |
0.0 |
|
|
|
|
|
min1 |
-421.77051 |
-1.39 |
|
|
|
|
|
sp2 |
-421.60813 |
-0.68 |
|
|
|
|
|
cluster2
+ C2 |
-193.87682 |
|
|
glyoxal |
-227.73297 |
|
|
|
-421.60979 |
-0.69 |
a Energy difference (aJ).
Table VIb. Cluster + C2tool
Energetics.
|
structure |
DFT |
DEa |
|
cluster
+ H |
-352.38179 |
|
|
c2tool |
-303.70708 |
|
|
|
-656.08887 |
0.0 |
|
|
|
|
|
min1 |
-656.38747 |
-1.30 |
|
|
|
|
|
cluster
+H + C2 |
-428.50013 |
|
|
glyoxal |
-227.73297 |
|
|
|
-656.23310 |
-0.63 |
a Energy difference (aJ).
Table VII. Cluster2 + C2
+ C4H2 Energetics.
|
structure |
DFT |
DEa |
|
cluster2
+ C2 |
-193.876819 |
|
|
C4H2 |
-153.32306 |
|
|
|
-347.19988 |
0.0 |
|
|
|
|
|
min1 |
-347.35530 |
-0.68 |
|
min2 |
-347.26911 |
-0.30 |
|
|
|
|
|
cluster2
+ C3 |
-231.94487 |
|
|
C3H2 |
-115.30456 |
|
|
|
-347.24943 |
-0.22 |
|
|
|
|
|
min3 |
-347.34152 |
-0.62 |
a Energy difference (aJ).
Table VIII. Cluster + C3.
|
structure |
DFT |
DEa |
|
min2 |
-465.89186 |
0.0 |
|
sp1 |
-465.87846 |
0.06 |
|
min1 |
-465.91599 |
-0.10 |
a Energy difference (aJ).
Table IX. Energetics
for diamond 100 + carbene tool.
|
structure |
DFT |
DEa |
|
|
|
|
|
cluster100 |
-349.90617 |
|
|
carbene
tool |
-153.32306 |
|
|
|
-503.22923 |
0.0 |
|
|
|
|
|
min1 |
-503.41654 |
-0.82 |
|
|
|
|
|
twisted |
-503.31265 |
-0.36 |
|
|
|
|
|
cluster100+c |
-387.89108 |
|
|
C3H2 |
-115.30456 |
|
|
|
-503.19564 |
0.15 |
a Energy difference (aJ).